Travaglini, K.J., Nabhan, A.N., Penland, L. et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature, 587(7835), 619–625. DOI: 10.1038/s41586-020-2922-4
Villani, A.-C., Satija, R., Reynolds, G. et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science, 356(6335), eaah4573. DOI: 10.1126/science.aah4573
Szabo, P.A., Levitin, H.M., Miron, M. et al. (2019). Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nature Communications, 10, 4706. DOI: 10.1038/s41467-019-12464-3
Bjorklund, A.K., Forkel, M., Picelli, S. et al. (2016). The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nature Immunology, 17(4), 451–460. DOI: 10.1038/ni.3368
Zheng, C., Zheng, L., Yoo, J.-K. et al. (2017). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell, 169(7), 1342–1356.e16. DOI: 10.1016/j.cell.2017.05.035
Cao, J., Spielmann, M., Qiu, X. et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature, 566(7745), 496–502. DOI: 10.1038/s41586-019-0969-x
Delgado, A.P. et al. (2022). Single-cell transcriptome analysis reveals evolutionarily conserved features during the transition from normal breast stromal cells to cancer-associated fibroblasts. bioRxiv (preprint). DOI: 10.1101/2022.05.05.490693
Jin, S., Guerrero-Juarez, C.F., Zhang, L. et al. (2021). Inference and analysis of cell-cell communication using CellChat. Nature Communications, 12, 1088. DOI: 10.1038/s41467-021-21246-9
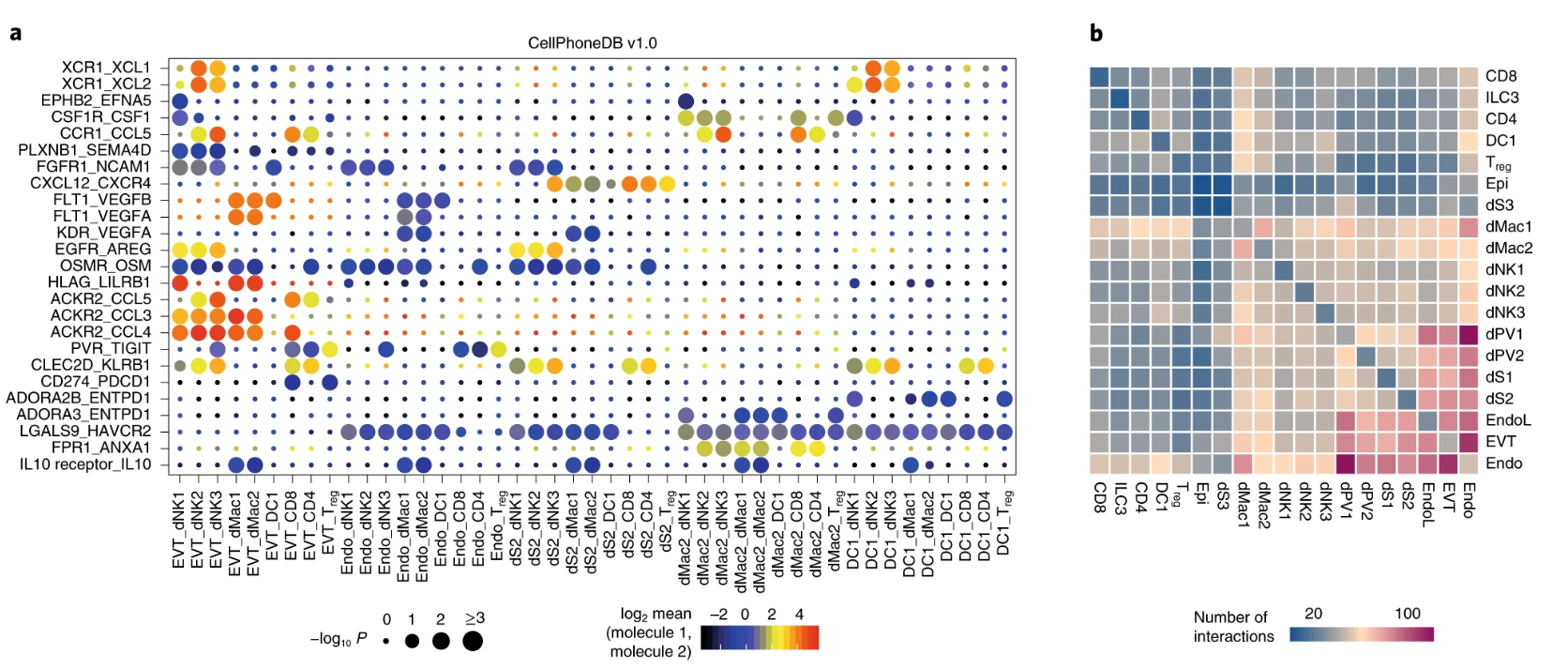
Efremova, M., Vento-Tormo, M., Teichmann, S.A. & Vento-Tormo, R. (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nature Protocols, 15(4), 1484–1506. DOI: 10.1038/s41596-020-0292-x
Tang, F., Barbacioru, C., Wang, Y. et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6(5), 377–382. DOI: 10.1038/nmeth.1315
Zheng, G.X.Y., Terry, J.M., Belgrader, P. et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications, 8, 14049. DOI: 10.1038/ncomms14049
Svensson, V., Vento-Tormo, R. & Teichmann, S.A. (2018). Exponential scaling of single-cell RNA-seq in the past decade. Nature Protocols, 13(4), 599–604. DOI: 10.1038/nprot.2017.149
Hao, Y., Stuart, T., Kowalski, M.H. et al. (2024). Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nature Biotechnology, 42(2), 293–304. DOI: 10.1038/s41587-023-01767-y
Wolf, F.A., Angerer, P. & Theis, F.J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology, 19, 15. DOI: 10.1186/s13059-017-1382-0
Regev, A., Teichmann, S.A., Lander, E.S. et al. (2017). The Human Cell Atlas. eLife, 6, e27041. DOI: 10.7554/eLife.27041
The Tabula Sapiens Consortium, Jones, R.C., Karkanias, J. et al. (2022). The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science, 376(6594), eabl4896. DOI: 10.1126/science.abl4896
Abdulla, S., Aevermann, B., Assis, P. et al. (2025). CZ CELLxGENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Nucleic Acids Research, 53(D1), D886–D900. DOI: 10.1093/nar/gkae1142
Yost, K.E., Satpathy, A.T., Wells, D.K. et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature Medicine, 25, 1251–1259. DOI: 10.1038/s41591-019-0522-3
van den Brink, S.C., Sage, F., Vértesy, Á. et al. (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods, 14(10), 935–936. DOI: 10.1038/nmeth.4437
O’Flanagan, C.H., Campbell, K.R., Zhang, A.W. et al. (2019). Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biology, 20, 210. DOI: 10.1186/s13059-019-1830-0
Adam, M., Potter, A.S. & Potter, S.S. (2017). Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development, 144(19), 3625–3632. DOI: 10.1242/dev.151142
Denisenko, E., Guo, B.B., Jones, M. et al. (2020). Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology, 21, 130. DOI: 10.1186/s13059-020-02048-6
Tosti, L., Hang, Y., Debnath, O. et al. (2021). Single-Nucleus and In Situ RNA-Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology, 160(4), 1330–1344.e11. DOI: 10.1053/j.gastro.2020.11.010
Bakken, T.E., Hodge, R.D., Miller, J.A. et al. (2018). Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE, 13(12), e0209648. DOI: 10.1371/journal.pone.0209648
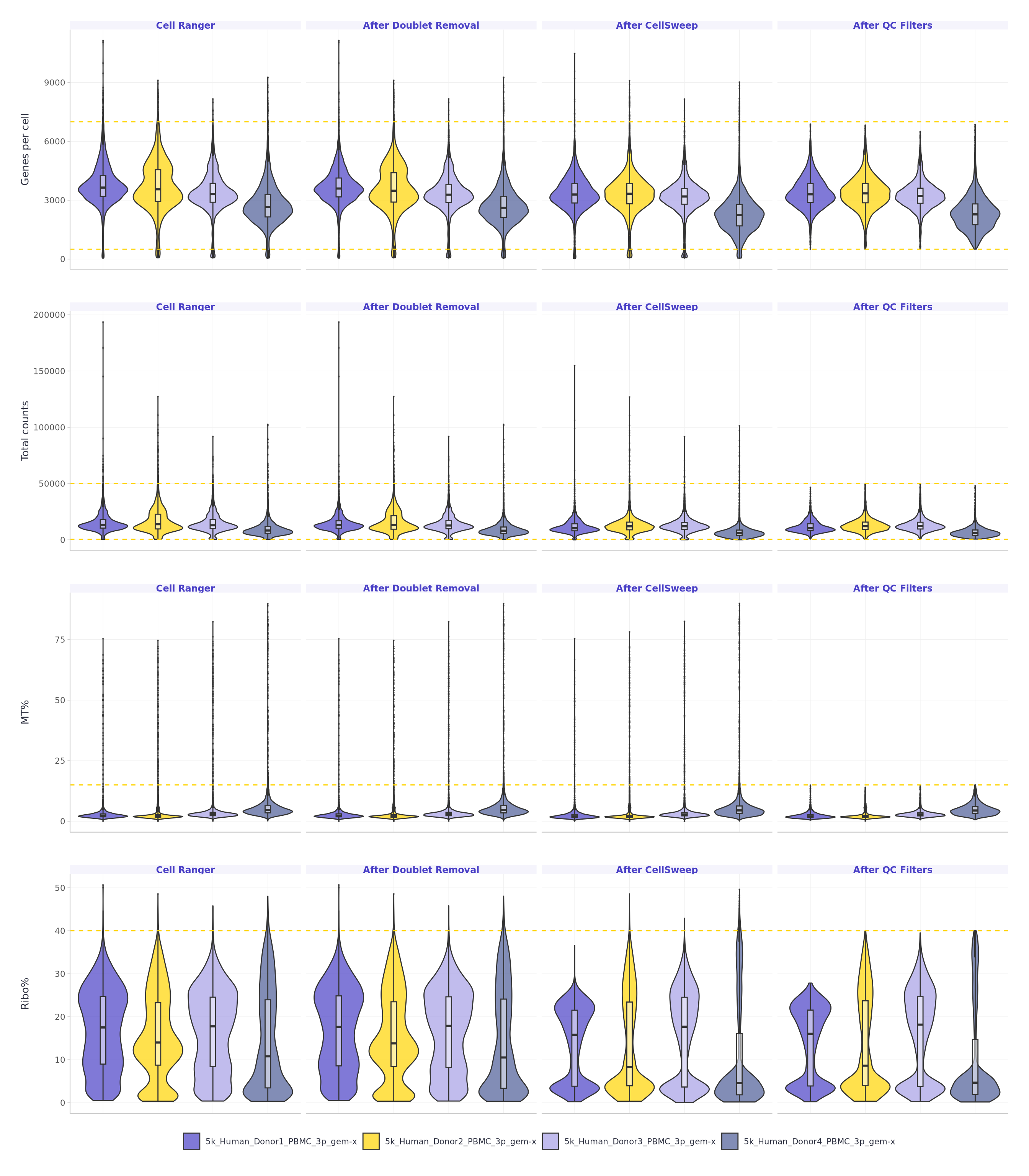
Lun, A.T.L., Riesenfeld, S., Andrews, T. et al. (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biology, 20, 63. DOI: 10.1186/s13059-019-1662-y
Fleming, S.J., Chaffin, M.D., Arduini, A. et al. (2023). Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nature Methods, 20(9), 1323–1335. DOI: 10.1038/s41592-023-01943-7
Young, M.D. & Behjati, S. (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience, 9(12), giaa151. DOI: 10.1093/gigascience/giaa151
Yang, S., Corbett, S.E., Bhoj, V. et al. (2020). Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biology, 21, 57. DOI: 10.1186/s13059-020-1950-6
Caskey, M., Rich, J., Weber, R., Mortazavi, A., Pachter, L. & Hallgrimsdottir, I. (2026). Single-Cell Genomics Decontamination with CellSweep. bioRxiv (preprint). DOI: 10.64898/2026.03.04.709349
Wolock, S.L., Lopez, R. & Klein, A.M. (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Systems, 8(4), 281–291.e9. DOI: 10.1016/j.cels.2018.11.005
Hao, Y., Hao, S., Andersen-Nissen, E. et al. (2021). Integrated analysis of multimodal single-cell data. Cell, 184(13), 3573–3587.e29. DOI: 10.1016/j.cell.2021.04.048
Domínguez Conde, C., Xu, C., Jarvis, L.B. et al. (2022). Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science, 376(6594), eabl5197. DOI: 10.1126/science.abl5197
Zhang, X., Lan, Y., Xu, J. et al. (2023). CellMarker: a manually curated resource for comprehensively collecting cell markers. Nucleic Acids Research, 51(D1), D1007–D1015. DOI: 10.1093/nar/gkac947
Franzén, O., Gan, L.-M. & Björkegren, J.L.M. (2019). PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database, 2019, baz046. DOI: 10.1093/database/baz046
Karlsson, M., Zhang, C., Méar, L. et al. (2021). A single-cell type transcriptomics map of human tissues. Science Advances, 7(31), eabh2169. DOI: 10.1126/sciadv.abh2169
Ianevski, A., Giri, A.K. & Aittokallio, T. (2022). Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nature Communications, 13, 1246. DOI: 10.1038/s41467-022-28803-w
Han, X., Zhou, Z., Fei, L. et al. (2020). Construction of a human cell landscape at single-cell level. Nature, 581, 303–309. DOI: 10.1038/s41586-020-2157-4
Ramachandran, P., Dobie, R., Wilson-Kanamori, J.R. et al. (2019). Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature, 575, 512–518. DOI: 10.1038/s41586-019-1631-3
Smillie, C.S., Biton, M., Ordovas-Montanes, J. et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell, 178(3), 714–730.e22. DOI: 10.1016/j.cell.2019.06.029
Habermann, A.C., Gutierrez, A.J., Bui, L.T. et al. (2020). Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Science Advances, 6(28), eaba1972. DOI: 10.1126/sciadv.aba1972
Mathys, H., Davila-Velderrain, J., Peng, Z. et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570, 332–337. DOI: 10.1038/s41586-019-1195-2
Liao, M., Liu, Y., Yuan, J. et al. (2020). Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nature Medicine, 26, 842–844. DOI: 10.1038/s41591-020-0901-9
Badia-i-Mompel, P., Vélez Santiago, J., Braunger, J. et al. (2022). decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinformatics Advances, 2(1), vbac016. DOI: 10.1093/bioadv/vbac016
Garcia-Alonso, L., Holland, C.H., Ibrahim, M.M. et al. (2019). Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Research, 29(8), 1363–1375. DOI: 10.1101/gr.240663.118
Schubert, M., Klinger, B., Klünemann, M. et al. (2018). Perturbation-response genes reveal signaling footprints in cancer gene expression. Nature Communications, 9, 20. DOI: 10.1038/s41467-017-02391-6
Holland, C.H., Tanevski, J., Perales-Patón, J. et al. (2020). Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biology, 21, 36. DOI: 10.1186/s13059-020-1949-z
Andreatta, M. & Carmona, S.J. (2021). UCell: Robust and scalable single-cell gene signature scoring. Computational and Structural Biotechnology Journal, 19, 3796–3798. DOI: 10.1016/j.csbj.2021.06.043
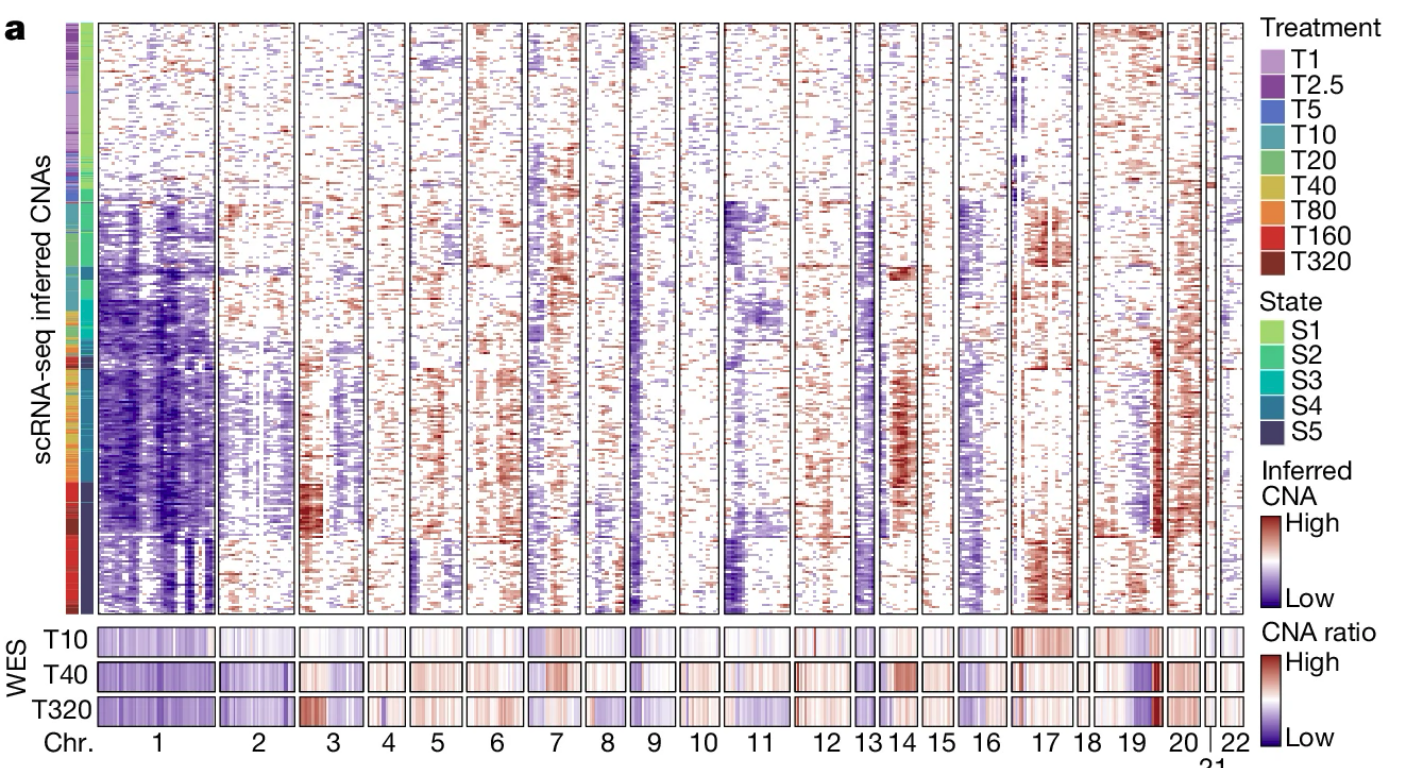
Patel, A.P., Tirosh, I., Trombetta, J.J. et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396–1401. DOI: 10.1126/science.1254257. This is the foundational study where the expression-based CNA-inference approach was first introduced and applied to glioblastoma, and on which inferCNV is based; inferCNV itself does not have a dedicated method paper. inferCNV repository: github.com/broadinstitute/infercnv. Recommended actively-maintained alternatives: infercna (github.com/jlaffy/infercna) and CopyKAT (see ref. [49]).
Gao, R., Bai, S., Henderson, Y.C. et al. (2021). Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nature Biotechnology, 39, 599–608. DOI: 10.1038/s41587-020-00795-2. CopyKAT repository: github.com/navinlabcode/copykat.
De Falco, A., Caruso, F., Su, X.D. et al. (2023). A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data. Nature Communications, 14, 1074. DOI: 10.1038/s41467-023-36790-9
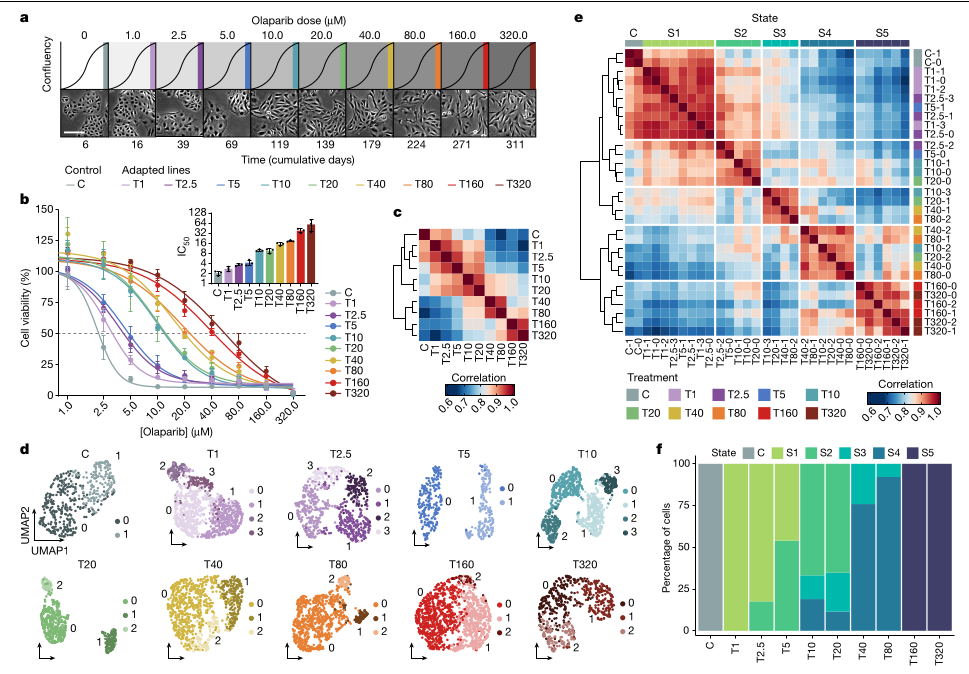
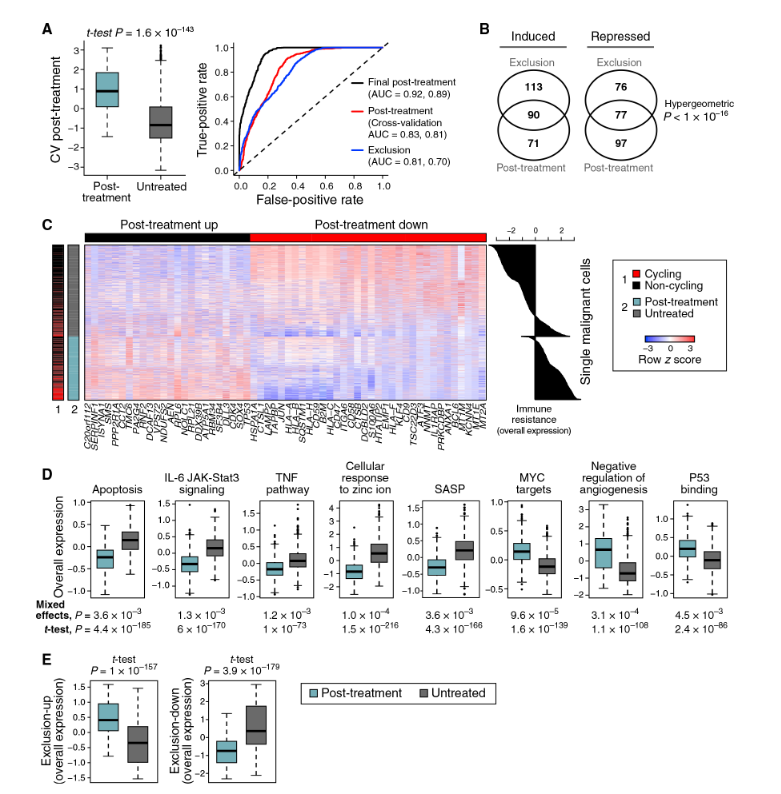
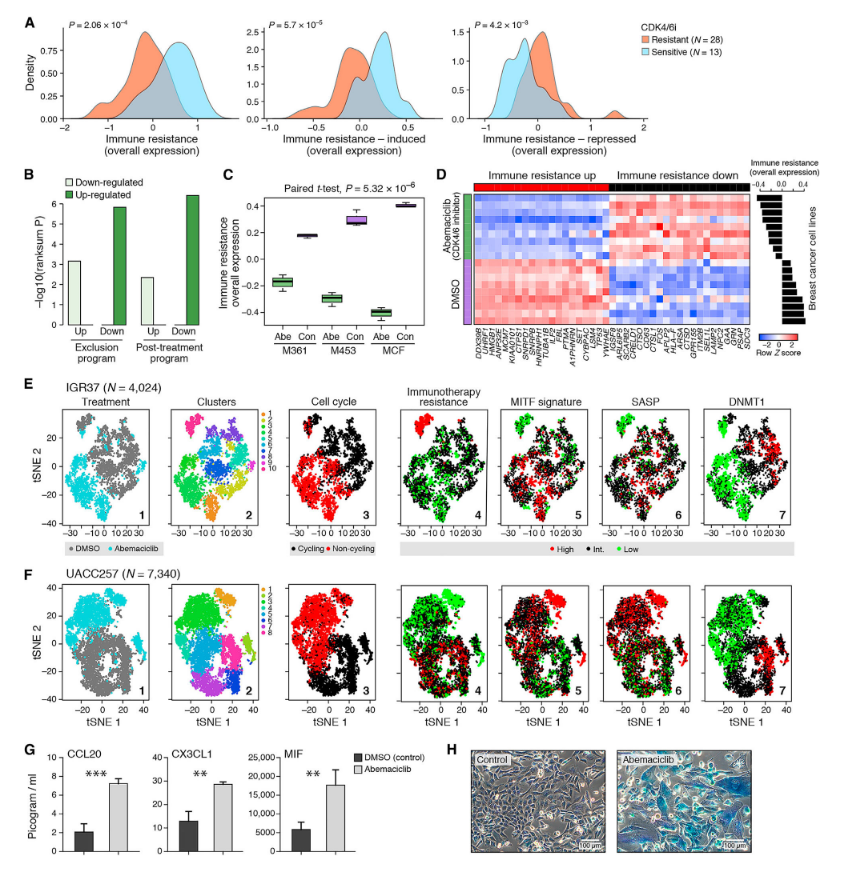
França, G.S., Baron, M., King, B.R. et al. (2024). Cellular adaptation to cancer therapy along a resistance continuum. Nature, 631, 876–883. DOI: 10.1038/s41586-024-07690-9
Vento-Tormo, R., Efremova, M., Botting, R.A. et al. (2018). Single-cell reconstruction of the early maternal-fetal interface in humans. Nature, 563, 347–353. DOI: 10.1038/s41586-018-0698-6
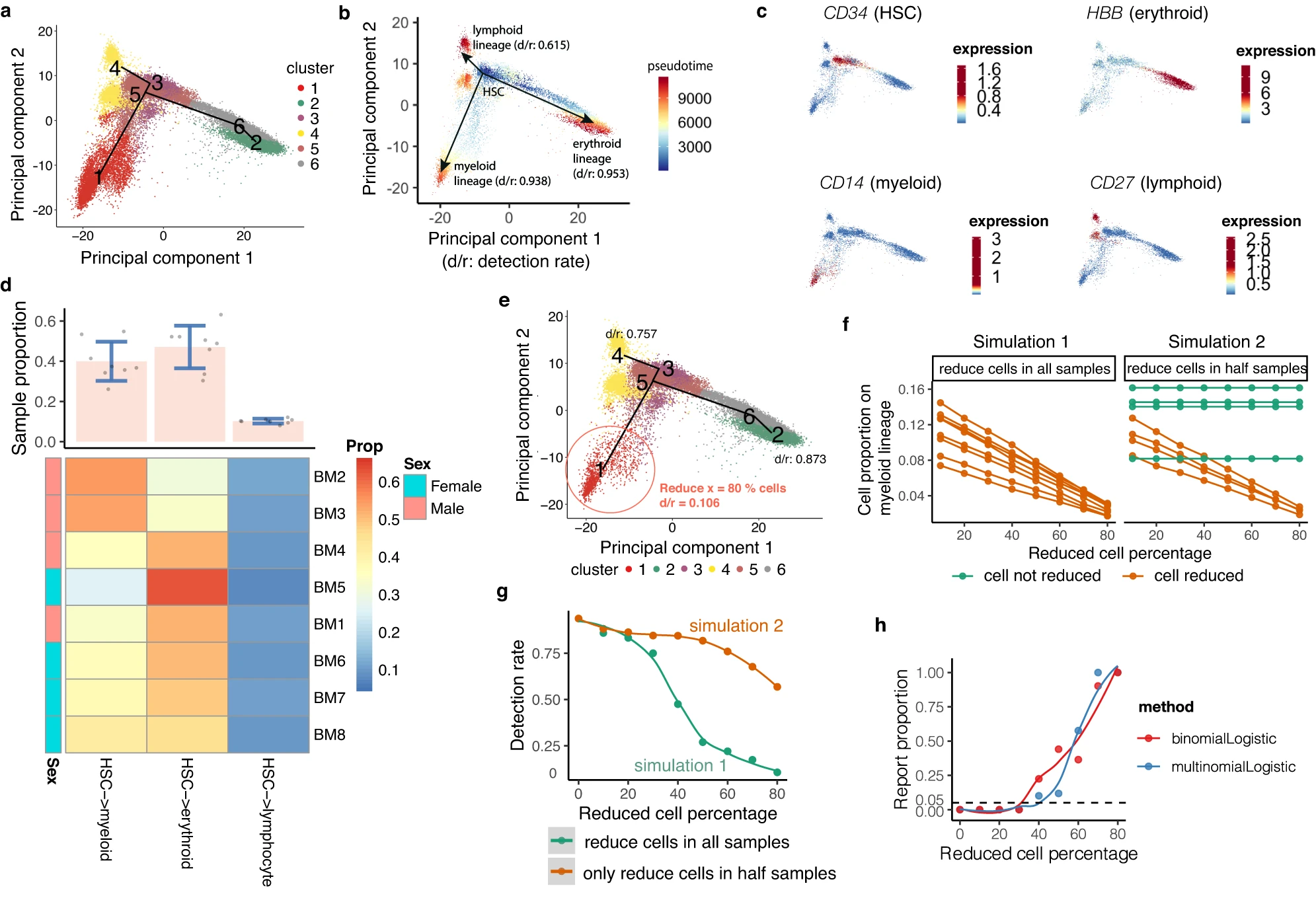
Street, K., Risso, D., Fletcher, R.B. et al. (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics, 19, 477. DOI: 10.1186/s12864-018-4772-0
La Manno, G., Soldatov, R., Zeisel, A. et al. (2018). RNA velocity of single cells. Nature, 560, 494–498. DOI: 10.1038/s41586-018-0414-6
Hou, W., Ji, Z., Chen, Z. et al. (2023). A statistical framework for differential pseudotime analysis with multiple single-cell RNA-seq samples. Nature Communications, 14, 7286. DOI: 10.1038/s41467-023-42841-y
Jerby-Arnon, L., Shah, P., Cuoco, M.S. et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell, 175(4), 984–997.e24. DOI: 10.1016/j.cell.2018.09.006
Martin, J.C., Chang, C., Boschetti, G. et al. (2019). Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell, 178(6), 1493–1508.e20. DOI: 10.1016/j.cell.2019.08.008
Rambow, F., Rogiers, A., Marin-Bejar, O. et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell, 174(4), 843–855.e19. DOI: 10.1016/j.cell.2018.06.025
Fustero-Torre, C., Jiménez-Santos, M.J., García-Martín, S., Carretero-Puche, C., García-Jimeno, L., Ivanchuk, V., Di Domenico, T., Gómez-López, G. & Al-Shahrour, F. (2021). Beyondcell: targeting cancer therapeutic heterogeneity in single-cell RNA sequencing data. Genome Medicine, 13, 187. DOI: 10.1186/s13073-021-00978-9
He, B., Xiao, Y., Liang, H. et al. (2023). ASGARD is A Single-cell Guided Pipeline to Aid Repurposing of Drugs. Nature Communications, 14, 993. DOI: 10.1038/s41467-023-36637-3
Pellecchia, S., Viscido, G., Franchini, M. & Gambardella, G. (2023). Predicting drug response from single-cell expression profiles of tumours. BMC Medicine, 21, 476. DOI: 10.1186/s12916-023-03182-1
Gustafsson, J., Held, F., Robinson, J.L. et al. (2024). scDrugPrio: a framework for the analysis of single-cell transcriptomics to address multiple problems in precision medicine in immune-mediated inflammatory diseases. Genome Medicine, 16, 42. DOI: 10.1186/s13073-024-01314-7
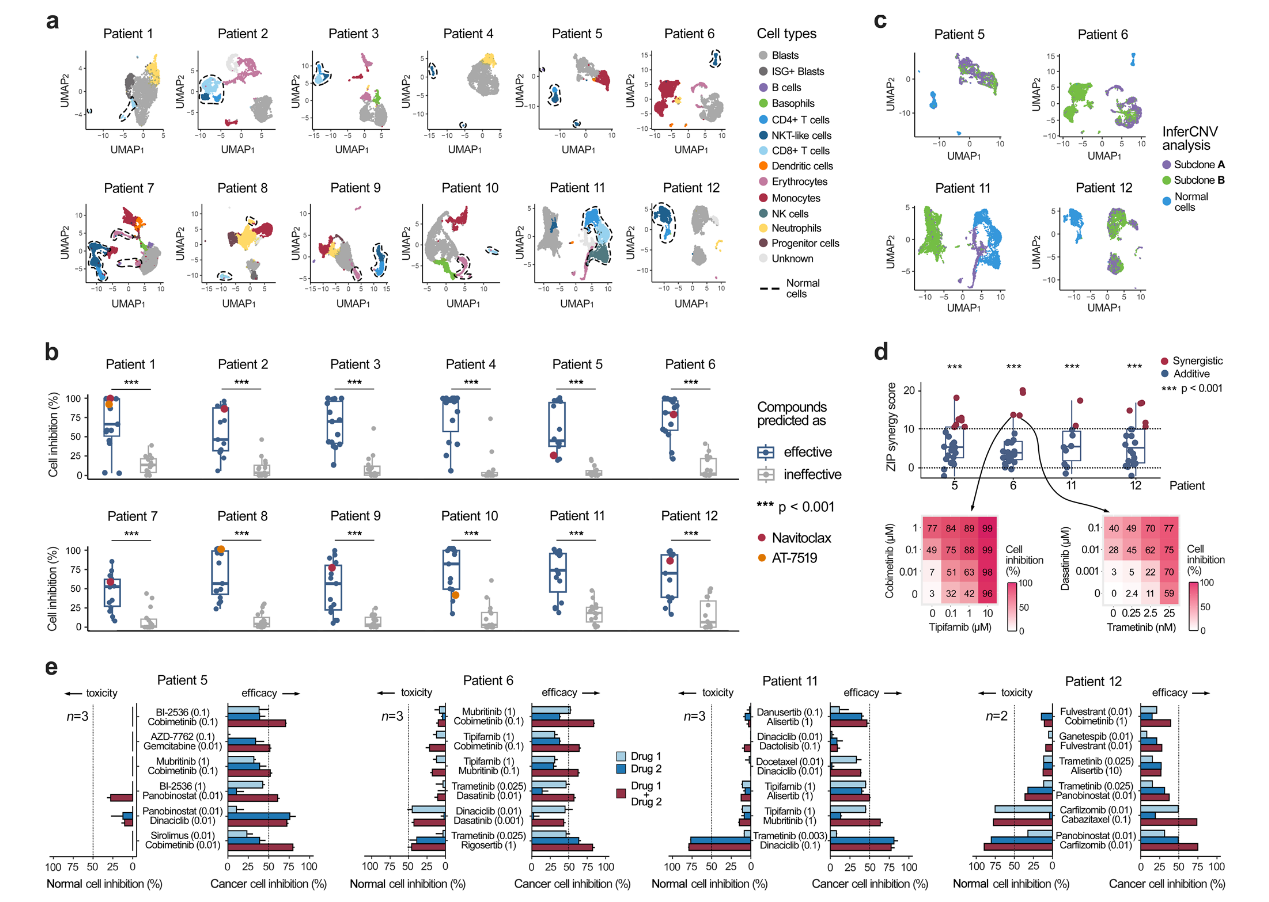
Ianevski, A., Nader, K., Driva, K. et al. (2024). Single-cell transcriptomes identify patient-tailored therapies for selective co-inhibition of cancer clones. Nature Communications, 15, 8579. DOI: 10.1038/s41467-024-52980-5
Lotfollahi, M., Wolf, F.A. & Theis, F.J. (2019). scGen predicts single-cell perturbation responses. Nature Methods, 16, 715–721. DOI: 10.1038/s41592-019-0494-8
Lotfollahi, M., Klimovskaia Susmelj, A., De Donno, C. et al. (2023). Predicting cellular responses to complex perturbations in high-throughput screens. Molecular Systems Biology, 19, e11517. DOI: 10.15252/msb.202211517
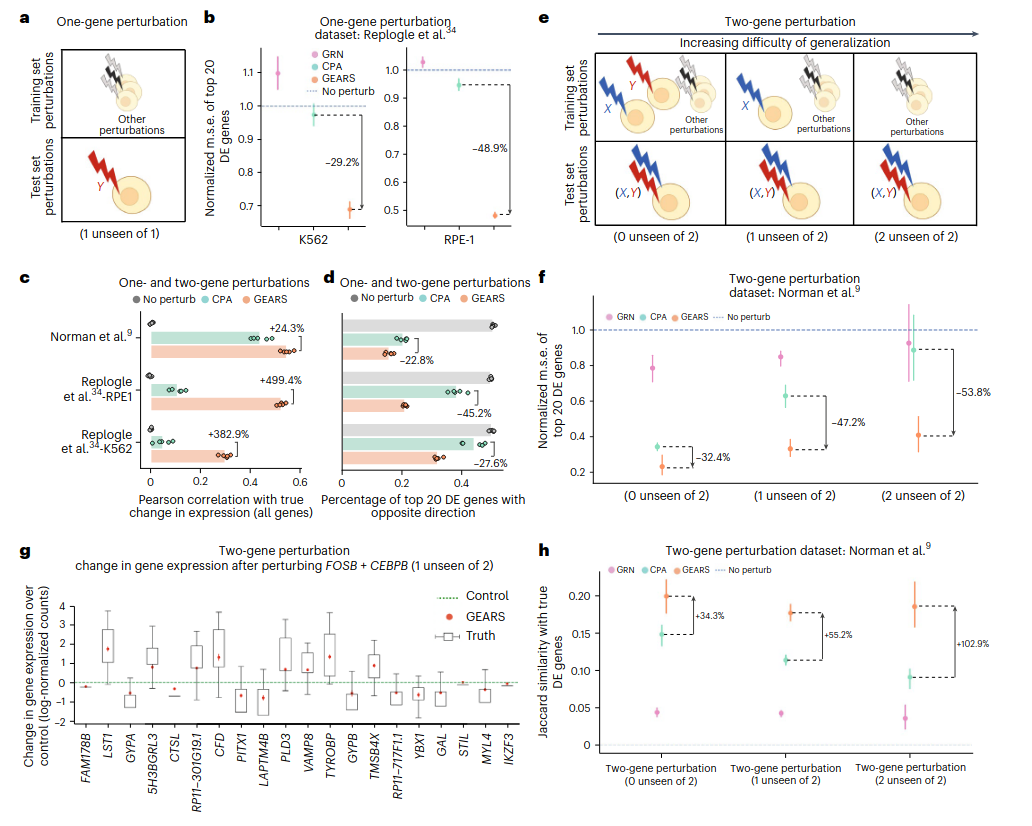
Roohani, Y., Huang, K. & Leskovec, J. (2024). Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nature Biotechnology, 42, 927–935. DOI: 10.1038/s41587-023-01905-6
Kernfeld, E.M., Keener, R. & Garmire, L.X. (2025). Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines. Nature Methods. DOI: 10.1038/s41592-025-02772-6
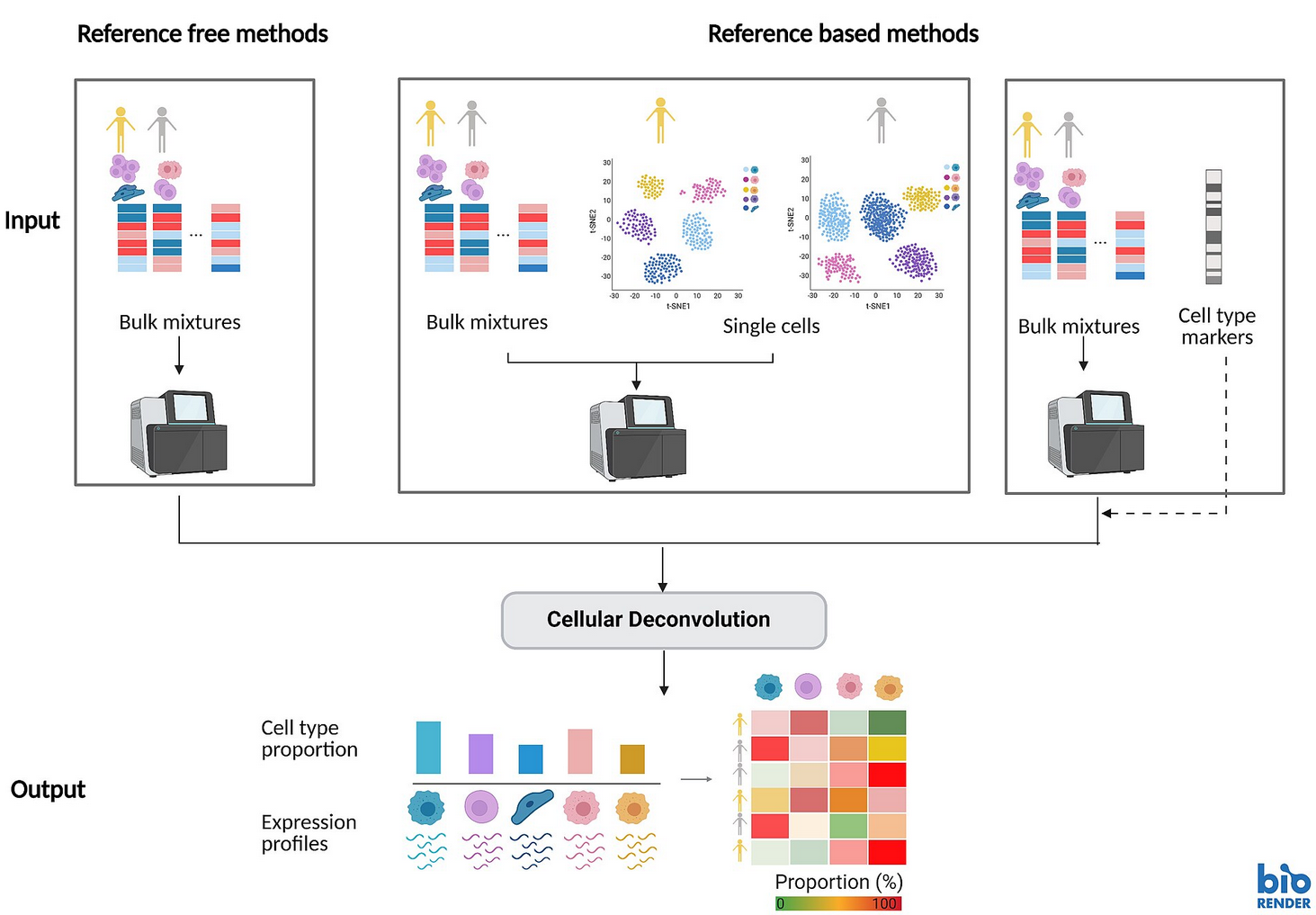
Tsoucas, D., Dong, R., Chen, H. et al. (2019). Accurate estimation of cell-type composition from gene expression data. Nature Communications, 10, 2975. DOI: 10.1038/s41467-019-10802-z
Chu, T., Wang, Z., Pe’er, D. & Bhatt, P. (2022). Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nature Cancer, 3, 505–517. DOI: 10.1038/s43018-022-00356-3
Xu, X., Li, R., Mo, O. et al. (2025). Cell-type deconvolution for bulk RNA-seq data using single-cell reference: a comparative analysis and recommendation guideline. Briefings in Bioinformatics, 26(1), bbaf031. DOI: 10.1093/bib/bbaf031